Fibrodisplasia Osificante Progresiva (FOP)
agosto 2009
VIVIR CON UNA ENFERMEDAD RARA
Manuel vive en Argentina. 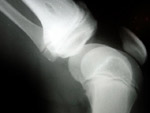 Tenía sólo 4 años cuando le diagnosticaron Fibrodisplasia Osificante Progresiva (FOP), un desorden genético muy raro que afecta de 1 a 2 millones de personas. FOP es un desorden en el que se forma hueso en los músculos, tendones, ligamentos y otros tejidos conectivos, un fenómeno muy raro. Puentes de hueso extra se desarrollan a través de las articulaciones, restringiendo progresivamente el movimiento. FOP es una enfermedad en la que el cuerpo produce no sólo mucho hueso, sino un esqueleto extra. Manuel tuvo su primer brote en 2000, con 3 años. Un brote se produce cuando el cuerpo empieza a generar un hueso nuevo, lo que implica la inflamación del tejido. El proceso suele ser muy doloroso. Los primeros síntomas hacían sospechar un cáncer. ‘Le hicieron muchas pruebas a Manuel,’ explica su madre, Moira. ‘Le hicieron incluso una biopsia, que es un procedimiento incorrecto en FOP, porque una biopsia crea más hueso en el punto del impacto. A Manuel le diagnosticaron primero con miofibromatosis, luego con fasciitis. ¡Empecé a mandar 800 correos electrónicos a hospitales de todo el mundo con la descripción de los síntomas de mi hijo! Nadie había visto algo similar, así que empecé a pensar que el diagnóstico inicial no era correcto. Una de las respuestas que recibí sugería el nombre de un médico en Argentina, quien por fin pudo dar el diagnóstico correcto en marzo 2001.’
Tenía sólo 4 años cuando le diagnosticaron Fibrodisplasia Osificante Progresiva (FOP), un desorden genético muy raro que afecta de 1 a 2 millones de personas. FOP es un desorden en el que se forma hueso en los músculos, tendones, ligamentos y otros tejidos conectivos, un fenómeno muy raro. Puentes de hueso extra se desarrollan a través de las articulaciones, restringiendo progresivamente el movimiento. FOP es una enfermedad en la que el cuerpo produce no sólo mucho hueso, sino un esqueleto extra. Manuel tuvo su primer brote en 2000, con 3 años. Un brote se produce cuando el cuerpo empieza a generar un hueso nuevo, lo que implica la inflamación del tejido. El proceso suele ser muy doloroso. Los primeros síntomas hacían sospechar un cáncer. ‘Le hicieron muchas pruebas a Manuel,’ explica su madre, Moira. ‘Le hicieron incluso una biopsia, que es un procedimiento incorrecto en FOP, porque una biopsia crea más hueso en el punto del impacto. A Manuel le diagnosticaron primero con miofibromatosis, luego con fasciitis. ¡Empecé a mandar 800 correos electrónicos a hospitales de todo el mundo con la descripción de los síntomas de mi hijo! Nadie había visto algo similar, así que empecé a pensar que el diagnóstico inicial no era correcto. Una de las respuestas que recibí sugería el nombre de un médico en Argentina, quien por fin pudo dar el diagnóstico correcto en marzo 2001.’
FOP tiene muchos aspectos adversos con otras enfermedades raras y muy raras: diagnóstico tardío y a veces erróneo, falta de tratamiento, poco conocimiento científico, y falta de concienciación de la enfermedad ent re los profesionales sanitarios. Poco después del diagnóstico, Moira y su esposo conocieron la Asociación Internacional de Fibrodisplasia Osificante Progresiva (IFOPA). ‘La asociación nos dio toda la información que necesitábamos. Tuvimos que aprender muchas cosas diferentes, como por ejemplo cómo ajustar el entorno de Manuel, y cómo hablar de la FOP con él y la familia… Nos sentimos afortunados de que esta comunidad tan activa hubiera conseguido tantos progresos importantes en los últimos 16 años en conocer la enfermedad,’ explica Moira. IFOPA tiene miembros en 50 países diferentes y su sitio web ofrece foros de debate en internet, donde la gente puede intercambiar información en inglés, español y portugués. Moira Liljesthröm continuó siendo un miembro activo de la comunidad FOP ayudando a crear el grupo FOP Latinoamérico (ALAFOP) en 2003, una red de unas 80 personas afectadas por la enfermedad de 10 países de Latinoamérica.
re los profesionales sanitarios. Poco después del diagnóstico, Moira y su esposo conocieron la Asociación Internacional de Fibrodisplasia Osificante Progresiva (IFOPA). ‘La asociación nos dio toda la información que necesitábamos. Tuvimos que aprender muchas cosas diferentes, como por ejemplo cómo ajustar el entorno de Manuel, y cómo hablar de la FOP con él y la familia… Nos sentimos afortunados de que esta comunidad tan activa hubiera conseguido tantos progresos importantes en los últimos 16 años en conocer la enfermedad,’ explica Moira. IFOPA tiene miembros en 50 países diferentes y su sitio web ofrece foros de debate en internet, donde la gente puede intercambiar información en inglés, español y portugués. Moira Liljesthröm continuó siendo un miembro activo de la comunidad FOP ayudando a crear el grupo FOP Latinoamérico (ALAFOP) en 2003, una red de unas 80 personas afectadas por la enfermedad de 10 países de Latinoamérica.
Recientemente, su marido y ella crearon la Fundación FOP en Argentina, que a la vez que trabaja en FOP está llevando a cabo un proyecto de investigación sobre la situación social, médica y legal de las personas afectadas de enfermedades raras en Argentina. Este proyecto está financiado por la Agencia Científica y Tecnológica del Gobierno Federal de Argentina. En lo que concierne al proyecto, Moira cree que Eurordis puede ayudar a la Fundación FOP. ‘Podríamos usar la experiencia de EURORDIS sobre cómo dirigir encuestas de investigación como las de EurordisCare 1 y EurordisCare 2. Creo que la cooperación entre redes, grupos y asociaciones nos ayudará a poner a las enfermedades raras en todas las agendas de salud pública y aumentar la concienc iación.’
iación.’
Una razón para no tirar la toalla es la esperanza del descubrimiento del gen de FOP en 2006 por investigadores de la Universidad de Pennsylvania School de Medicina. El descubrimiento del gen de FOP y la única mutación que causa FOP proporciona un objetivo específico para el futuro desarrollo de un medicamento, no sólo para tratar los síntomas, sino también la enfermedad. Esta investigación tuvo financiación en parte de IFOPA. Hoy, Manuel está en 5º y acude a la escuela con un entorno seguro que limita los traumas que pueda sufrir. ‘Manuel tiene dificultad para levantar los brazos y doblar la cintura, pero no le impide jugar y hacer muchas otras cosas’ dice su madre orgullosa.
Este artículo apareció previamente en el número de Junio de 2007 de nuestro boletín de noticias.
Autor: Nathacha Appanah
Traductor: Conchi Casas Jorde
Fotos: Todas las fotos © Liljesthröm exceptúan hueso © Clara Natoli