Fibrodysplasia Ossificans Progressiva (FOP)
August 2009LIVING WITH A RARE DISEASE
Fibrodysplasia Ossificans Progressiva (FOP)
Manuel and his family live in Argentina. He was only 4 years old when he 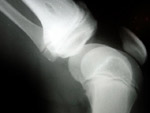 was diagnosed with Fibrodysplasia Ossificans Progressiva (FOP), an extremely rare genetic condition that affects 1 in 2 million people. FOP is a disorder in which bone is formed in muscles, tendons, ligaments, and other connective tissues, a very rare phenomenon. Bridges of extra bone form across the joints in characteristic patterns, progressively restricting movement. FOP is a disease in which the body produces not just too much bone, but an extra skeleton. Manuel had his first flare-up in 2000, at the age of 3. A flare-up occurs when the body starts to generate new bone, leading to tissue swelling. The process is usually very painful. His symptoms were first suspected as being the signs of cancer. ‘Manuel underwent many tests,’ explains Manuel’s mother, Moira. ‘A biopsy was even performed, which is a totally incorrect procedure in FOP, because the shock of the biopsy creates new bone at the point of impact. Manuel got an initial diagnosis of myofibromatosis, and then another one of fasciitis. I began to send 800 emails to hospitals all over the world with a description of my son’s symptoms!
was diagnosed with Fibrodysplasia Ossificans Progressiva (FOP), an extremely rare genetic condition that affects 1 in 2 million people. FOP is a disorder in which bone is formed in muscles, tendons, ligaments, and other connective tissues, a very rare phenomenon. Bridges of extra bone form across the joints in characteristic patterns, progressively restricting movement. FOP is a disease in which the body produces not just too much bone, but an extra skeleton. Manuel had his first flare-up in 2000, at the age of 3. A flare-up occurs when the body starts to generate new bone, leading to tissue swelling. The process is usually very painful. His symptoms were first suspected as being the signs of cancer. ‘Manuel underwent many tests,’ explains Manuel’s mother, Moira. ‘A biopsy was even performed, which is a totally incorrect procedure in FOP, because the shock of the biopsy creates new bone at the point of impact. Manuel got an initial diagnosis of myofibromatosis, and then another one of fasciitis. I began to send 800 emails to hospitals all over the world with a description of my son’s symptoms!  Nobody had ever seen anything similar, so we began to think that the diagnosis we got initially was wrong. One of the replies we received suggested the name of a doctor in Argentina, who was finally able to make the correct diagnosis, in March 2001.’
Nobody had ever seen anything similar, so we began to think that the diagnosis we got initially was wrong. One of the replies we received suggested the name of a doctor in Argentina, who was finally able to make the correct diagnosis, in March 2001.’
FOP shares many adverse features with other rare and very rare diseases: delayed and sometimes wrong diagnosis, no treatment, little scientific knowledge, and lack of awareness of the disease among health professionals. Soon after the diagnosis, Moira and her husband came across the International Fibrodysplasia Ossificans Progressiva Association (IFOPA). ‘The association gave us all the information we needed. We had to learn many different things, such as how to adjust the environment for Manuel, and how to speak about FOP with him and our family… We felt lucky that this pro-active community had made important progress in the last 16 years in getting to know the disease,’ explains Moira. The IFOPA has members in 50 different countries and its website offers internet discussion forums where people can exchange information in English, Spanish and Portuguese. Moira Liljesthröm went on to be an active member of the FOP community by helping to set up the Latino American FOP group (ALAFOP) in 2003, a network of around 80 people affected by the disease from 10 Latin-American countries. More recently, she and her husband set up Fundación FOP in Argentina, which, as well as working on FOP, is running a research project on the social, medical and legal situation of people affected by rare diseases in Argentina. This project is fun ded by the Scientific and Technological Agency of the Federal Government of Argentina. In regards to the project, Moira thinks Eurordis can help Fundación FOP. ‘We could really use EURORDIS’ experience on how to conduct research surveys, such as the EurordisCare 1 and EurordisCare 2 surveys. I think that cooperation between networks, groups, and associations will help to put rare diseases on every health public agenda and increase awareness.’
ded by the Scientific and Technological Agency of the Federal Government of Argentina. In regards to the project, Moira thinks Eurordis can help Fundación FOP. ‘We could really use EURORDIS’ experience on how to conduct research surveys, such as the EurordisCare 1 and EurordisCare 2 surveys. I think that cooperation between networks, groups, and associations will help to put rare diseases on every health public agenda and increase awareness.’
One reason not to give up hope is the discovery of the FOP gene in 2006 by researchers from the University of Pennsylvania School of Medicine. The discovery of the FOP gene and the unique mutation that causes FOP provides a highly specific target for future drug development, not just to treat the symptoms, but also the disease itself. This research was partly funded by IFOPA. Today, Manuel is in 5th grade and attends a school with a safe environment to limit the traumas he could suffer. ‘Manuel has some difficulties in raising his arms and bending his waist, but this doesn’t stop him from playing and doing many other things!’ says his proud mother.
This article was previously published in the June 2007 issue of our newsletter.
Author: Nathacha Appanah
Photos: all photos © Liljesthröm except bone © Clara Natoli